

BF-1 interferes with transforming growth factor beta signaling by associating with Smad partners
- PMID: 10938097
- PMCID: PMC86095
- DOI: 10.1128/MCB.20.17.6201-6211.2000
BF-1 interferes with transforming growth factor beta signaling by associating with Smad partners
Abstract
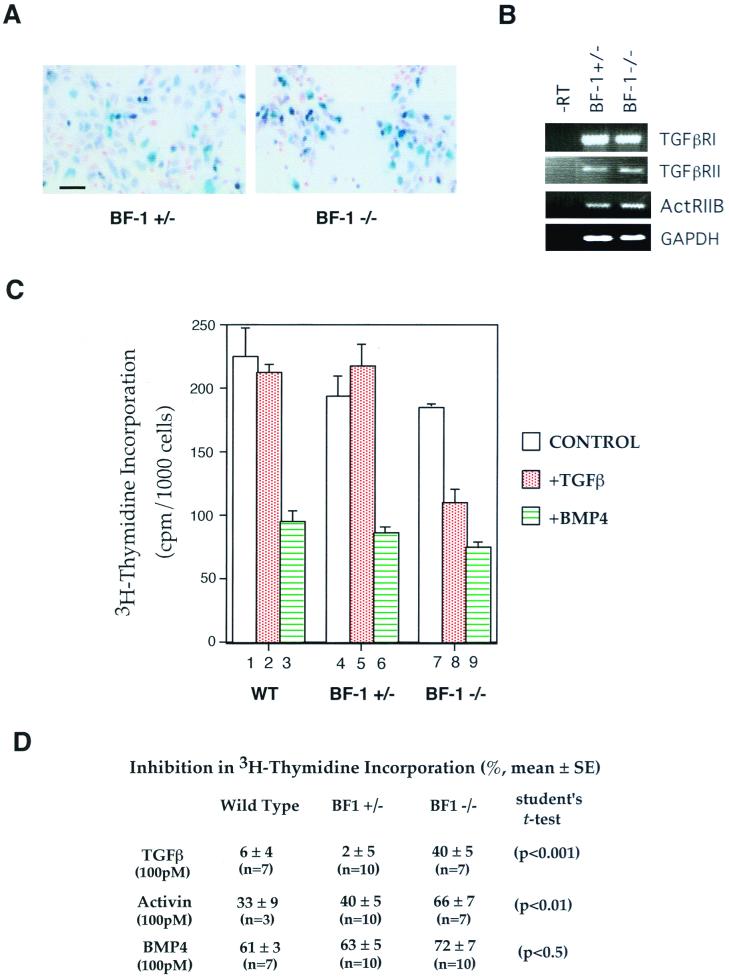
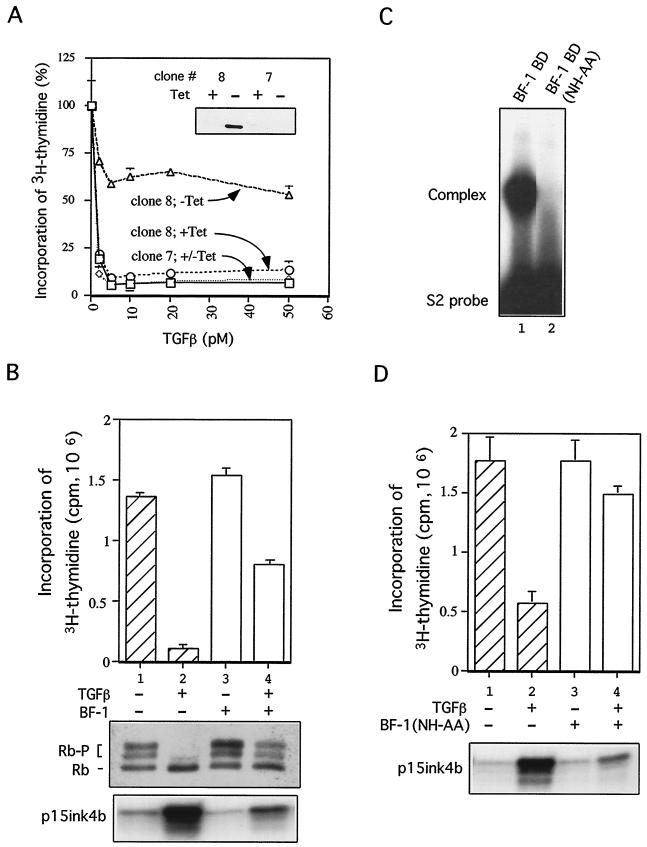
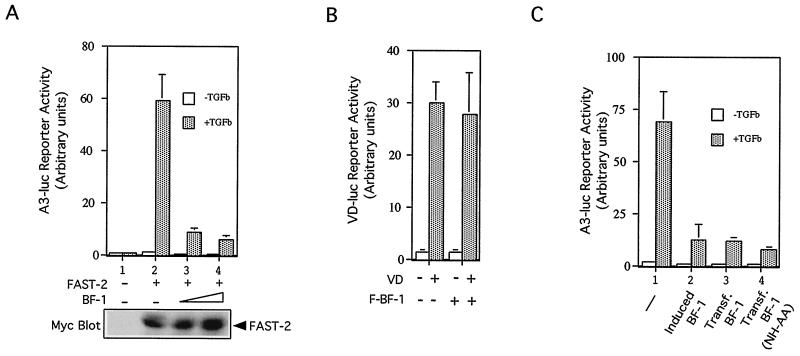
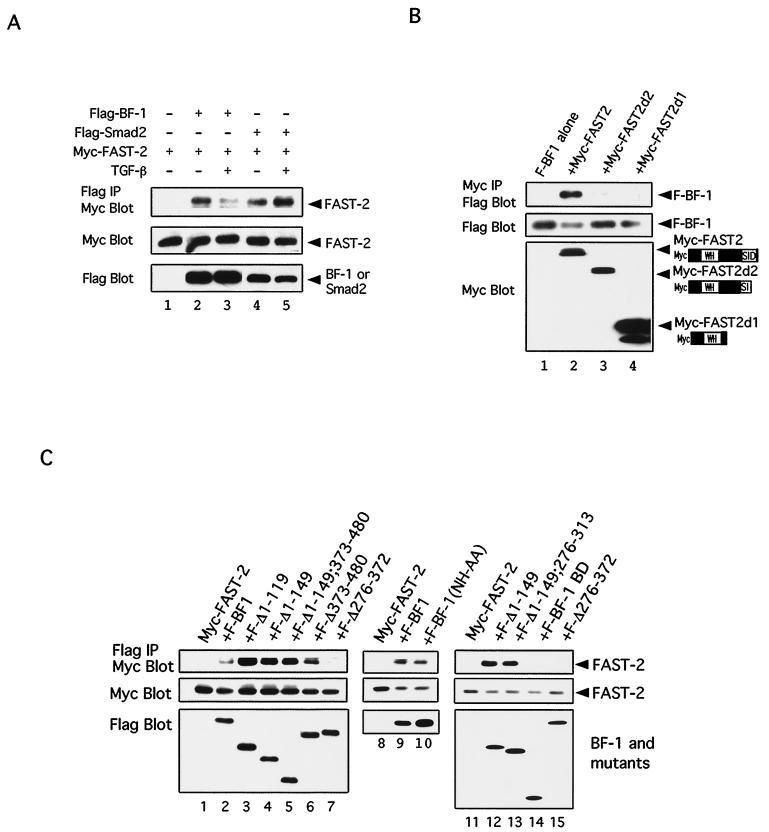
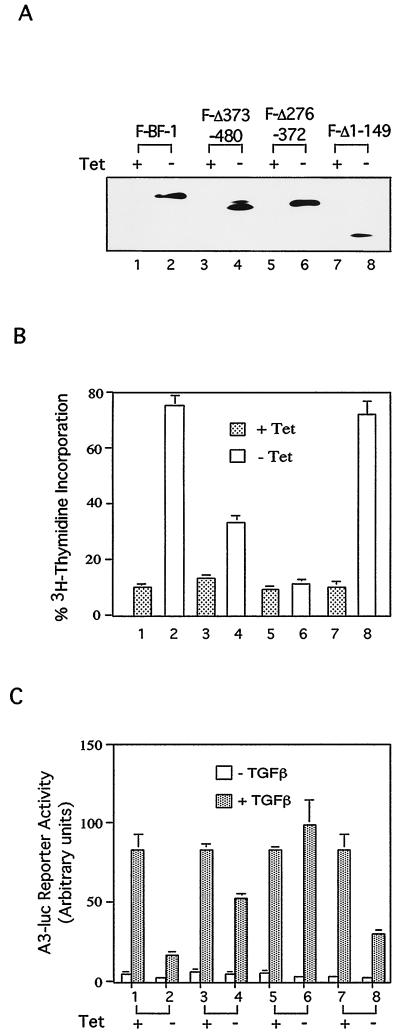
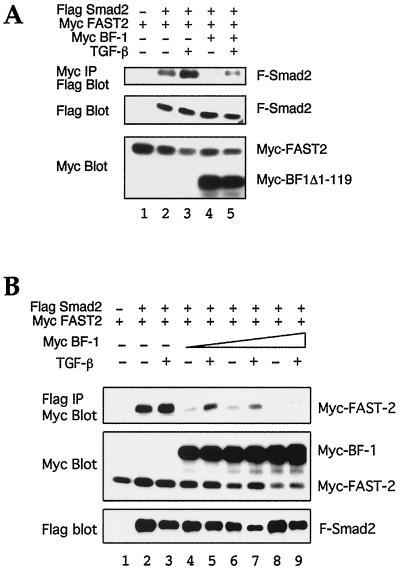
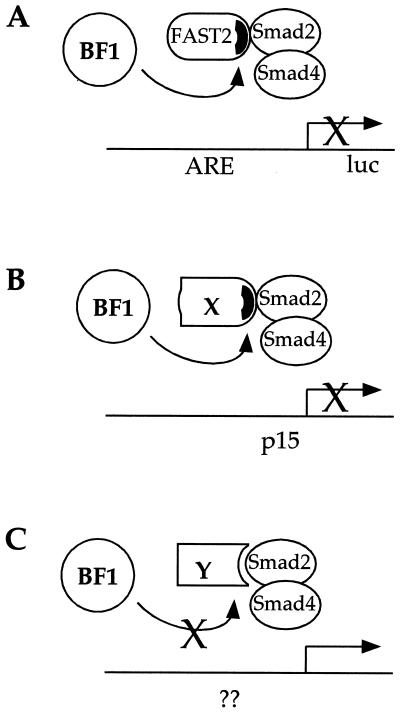
The winged-helix (WH) BF-1 gene, which encodes brain factor 1 (BF-1) (also known as foxg1), is essential for the proliferation of the progenitor cells of the cerebral cortex. Here we show that BF-1-deficient telencephalic progenitor cells are more apt to leave the cell cycle in response to transforming growth factor beta (TGF-beta) and activin. We found that ectopic expression of BF-1 in vitro inhibits TGF-beta mediated growth inhibition and transcriptional activation. Surprisingly, we found that the ability of BF-1 to function as a TGF-beta antagonist does not require its DNA binding activity. Therefore, we investigated whether BF-1 can inhibit Smad-dependent transcriptional responses by interacting with Smads or Smad binding partners. We found that BF-1 does not interact with Smads. Because the identities of the Smad partners mediating growth inhibition by TGF-beta are not clearly established, we examined a model reporter system which is known to be activated by activin and TGF-beta through Smads and the WH factor FAST-2. We demonstrate that BF-1 associates with FAST-2. This interaction is dependent on the same region of protein which mediates its ability to interfere with the antiproliferative activity of TGF-beta and with TGF-beta-dependent transcriptional activation. Furthermore, the interaction of FAST-2 with BF-1 is mediated by the same domain which is required for FAST-2 to interact with Smad2. We propose a model in which BF-1 interferes with transcriptional responses to TGF-beta by interacting with FAST-2 or with other DNA binding proteins which function as Smad2 partners and which have a common mode of interaction with Smad2.
Figures

Similar articles
-
Functional cloning of the proto-oncogene brain factor-1 (BF-1) as a Smad-binding antagonist of transforming growth factor-beta signaling.J Biol Chem. 2001 Aug 10;276(32):30224-30. doi: 10.1074/jbc.M102759200. Epub 2001 May 31. J Biol Chem. 2001. PMID: 11387330
-
Nuclear factor YY1 inhibits transforming growth factor beta- and bone morphogenetic protein-induced cell differentiation.Mol Cell Biol. 2003 Jul;23(13):4494-510. doi: 10.1128/MCB.23.13.4494-4510.2003. Mol Cell Biol. 2003. PMID: 12808092 Free PMC article.
-
The transcriptional co-activator P/CAF potentiates TGF-beta/Smad signaling.Nucleic Acids Res. 2000 Nov 1;28(21):4291-8. doi: 10.1093/nar/28.21.4291. Nucleic Acids Res. 2000. PMID: 11058129 Free PMC article.
-
The transcriptional role of Smads and FAST (FoxH1) in TGFbeta and activin signalling.Mol Cell Endocrinol. 2001 Jun 30;180(1-2):3-11. doi: 10.1016/s0303-7207(01)00524-x. Mol Cell Endocrinol. 2001. PMID: 11451566 Review.
-
Signal transduction of the TGF-beta superfamily by Smad proteins.J Biochem. 1999 Jan;125(1):9-16. doi: 10.1093/oxfordjournals.jbchem.a022273. J Biochem. 1999. PMID: 9880789 Review.
Cited by
-
In control of biology: of mice, men and Foxes.Biochem J. 2006 Jul 15;397(2):233-46. doi: 10.1042/BJ20060387. Biochem J. 2006. PMID: 16792526 Free PMC article. Review.
-
FOXG1 dose tunes cell proliferation dynamics in human forebrain progenitor cells.Stem Cell Reports. 2022 Mar 8;17(3):475-488. doi: 10.1016/j.stemcr.2022.01.010. Epub 2022 Feb 10. Stem Cell Reports. 2022. PMID: 35148845 Free PMC article.
-
Transcriptional control of embryonic and adult neural progenitor activity.Front Neurosci. 2023 Jul 28;17:1217596. doi: 10.3389/fnins.2023.1217596. eCollection 2023. Front Neurosci. 2023. PMID: 37588515 Free PMC article. Review.
-
FOXG1 targets BMP repressors and cell cycle inhibitors in human neural progenitor cells.Hum Mol Genet. 2023 Jul 20;32(15):2511-2522. doi: 10.1093/hmg/ddad089. Hum Mol Genet. 2023. PMID: 37216650 Free PMC article.
-
FoxG1 promotes the survival of postmitotic neurons.J Neurosci. 2011 Jan 12;31(2):402-13. doi: 10.1523/JNEUROSCI.2897-10.2011. J Neurosci. 2011. PMID: 21228151 Free PMC article.
References
-
- Caviness V J, Takahashi T, Nowakowski R. Numbers, time and neocortical neuronogenesis: a general developmental and evolutionary model. Trends Neurosci. 1995;18:379–383. - PubMed
-
- Chen X, Rubock M, Whitman M. A transcriptional partner for MAD proteins in TGF-B signalling. Nature. 1996;383:691–696. - PubMed
-
- Chen X, Weisberg E, Fridmacher V, Watanabe M, Naco G, Whitman M. Smad4 and FAST-1 in the assembly of activin-responsive factor. Nature. 1997;389:85–89. - PubMed
-
- Clark K, Halay E, Lai E, Burley S. Cocrystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature. 1993;364:412–420. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous